

Designing Well-Structured Cyclic Pentapeptides Based on Sequence–Structure Relationships
Designing Well-Structured Cyclic Pentapeptides Based on Sequence–Structure Relationships
J. Phys. Chem. B, 2018, 122 (14), pp 3908–3919
DOI: 10.1021/acs.jpcb.8b01747
Publication Date (Web): March 28, 2018
Diana P. Slough, Sean M. McHugh, Ashleigh E. Cummings, Peng Dai, Bradley L. Pentelute, Joshua A. Kritzer, and Yu-Shan Lin
Abstract
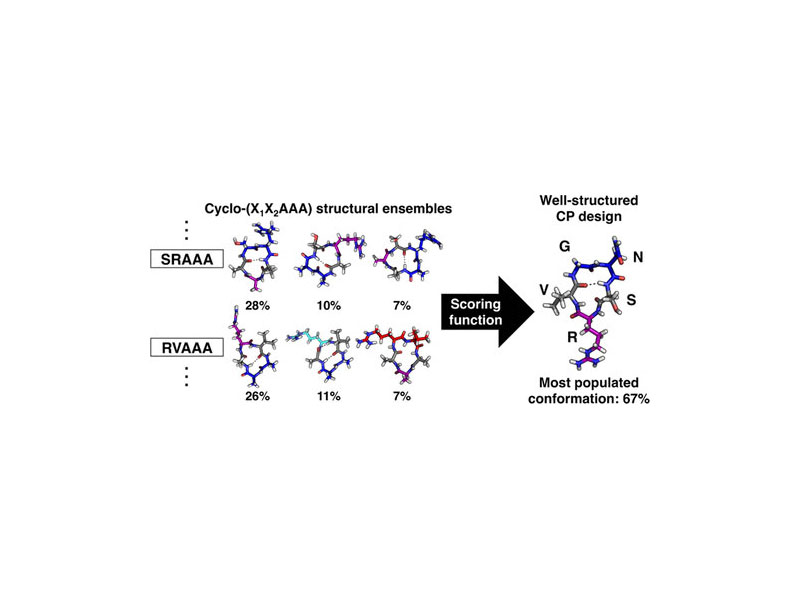
Cyclic peptides are a promising class of molecules for unique applications. Unfortunately, cyclic peptide design is severely limited by the difficulty in predicting the conformations they will adopt in solution. In this work, we use explicit-solvent molecular dynamics simulations to design well-structured cyclic peptides by studying their sequence–structure relationships. Critical to our approach is an enhanced sampling method that exploits the essential transitional motions of cyclic peptides to efficiently sample their conformational space. We simulated a range of cyclic pentapeptides from all-glycine to a library of cyclo-(X1X2AAA) peptides to map their conformational space and determine cooperative effects of neighboring residues. By combining the results from all cyclo-(X1X2AAA) peptides, we developed a scoring function to predict the structural preferences for X1–X2 residues within cyclic pentapeptides. Using this scoring function, we designed a cyclic pentapeptide, cyclo-(GNSRV), predicted to be well structured in aqueous solution. Subsequent circular dichroism and NMR spectroscopy revealed that this cyclic pentapeptide is indeed well structured in water, with a nuclear Overhauser effect and J-coupling values consistent with the predicted structure.